Cyclin D1, a classic proto-oncogene, is significantly upregulated in various cancers, including breast cancer, mantle cell lymphoma (MCL), bladder cancer, prostate cancer, non-small cell lung cancer, and colon cancer. However, it remains unclear in which cancer Cyclin D1 acts as the initiating driver.
Recently, the team led by Chair Professor Chen Di from Shenzhen University of Advanced Technology and Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences, revealed the critical role of Cyclin D1 in MCL pathogenesis. They found that Cyclin D1 homeostasis is co-regulated by phosphorylation- and SUMOylation-mediated proteasomal degradation. The findings were published online in Acta Pharmaceutica Sinica B (IF = 14.50) under the title “Disruption of cyclin D1 degradation leads to the development of mantle cell lymphoma.” Associate researcher Lu Ke from Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences, is the first author, and Chen Di is the corresponding author.
Cyclin D1 is a protein that works with CDK4/6 to regulate the cell cycle. Cyclin D1 levels are often elevated in many cancers. The researchers discovered that Cyclin D1 is jointly controlled by two protein degradation pathways: phosphorylation and SUMOylation. Disruption of both degradation pathways in mice led to an MCL-like phenotype. This indicates that Cyclin D1 plays a pivotal role in MCL development.
They also found that arsenic trioxide (ATO) enhances Cyclin D1 SUMOylation, promoting its degradation and inducing apoptosis in MCL cells. This suggests that ATO, already used for acute promyelocytic leukemia, may serve as a potential therapeutic agent for MCL.
Overall, this study provides new insights into the molecular mechanisms of MCL and offers novel strategies for its future treatment.
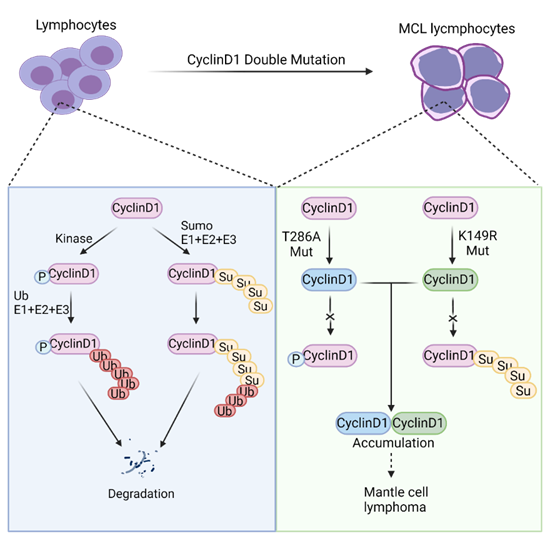
Figure 1: Cyclin D1 protein levels are controlled by phosphorylation- and SUMOylation-mediated proteasomal degradation. Inhibition of endogenous Cyclin D1 degradation leads to B-cell transformation and development of an MCL-like phenotype.
 CN
CN